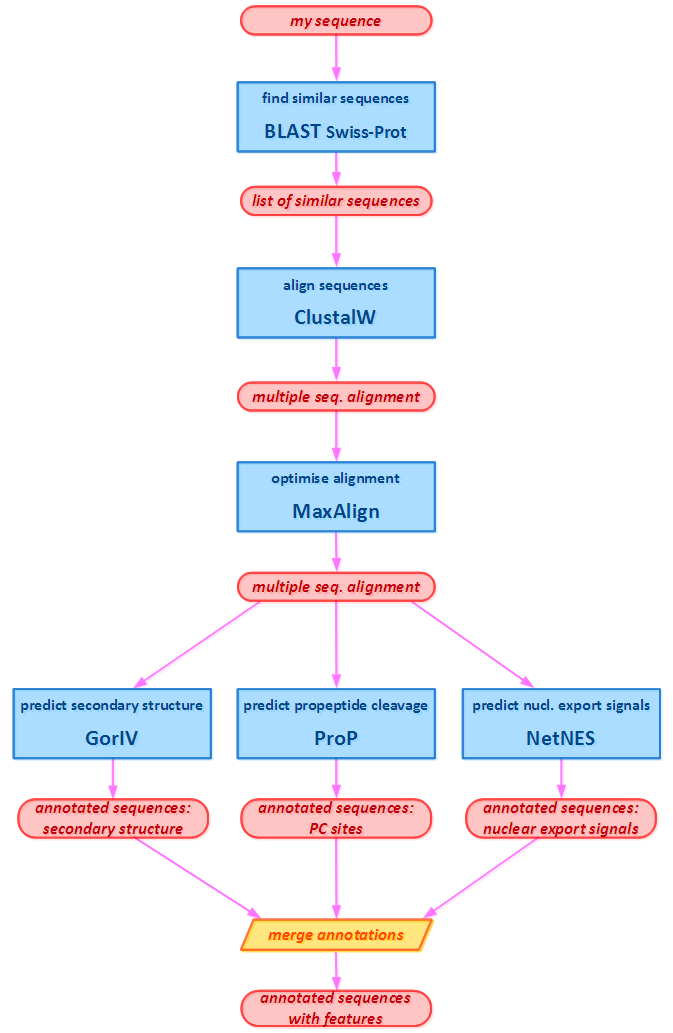
An example workflow combining multiple bioinformatics Web services hosted by multiple service providers. In the schematic figure, the blue rectangles are Web-service calls, the red ovals are data. The workflow does a routine bioinformatics analysis: Having a query amino-acid sequence of our interest, similar sequences are searched and fetched from a database. These sequences are then aligned into a multiple sequence alignment (which is in this example further optimised by MaxAlign). Annotations by various types of features, according to the particular interest, are fetched from multiple sources or predicted using multiple prediction tools. In this example, 3 different types of features are predicted: the secondary structure using GorIV, propeptide cleavage sites using ProP, and nuclear export signals using NetNES. These annotations are then merged into a single data object containing all the features of interest.
Java implementation:
WorkflowNr1.java The source code of this workflow in Java (fully in 1 main method), using Axis2 framework for calling Web services and XMLBeans for data binding.
Although the source code uses the very verbose XMLBeans data binding and contains a large portion of exception & special-case handling,
the data flow itself is otherwise very simple thanks to the use of common BioXSD formats of inputs and outputs of the included Web services.
WorkflowNr1.jar The executable Java archive.
[Run: "java -jar WorkflowNr1.jar"]
workflowNr1.zip The full Java project including all necessary libraries, the Axis2/XMLBeans-generated classes plus additional files, and Ant build file.
[Run/debug in Eclipse:
1. New>Project>Java Project from Existing Ant Buildfile>build.xml from the inflated workflowNr1 folder
| 2. src/org.bioxsd.workflows.WorkflowNr1.java: Run Configurations>Classpath>Advanced>Add Folders>workflowNr1/resources
| 3. Run/debug src/org.bioxsd.workflows.WorkflowNr1.java]
[Build WorkflowNr1.jar: Run "ant" in the inflated workflowNr1 folder]